After selecting a particular protein from the queue (or the database of repaired nontrivial proteins) users can view all information about it in the Results pages, which can be divided into following categories:

Fig. 1 Basic information about the job: project name, PDB ID (for structures from the PDB database), the unique job id and current status.
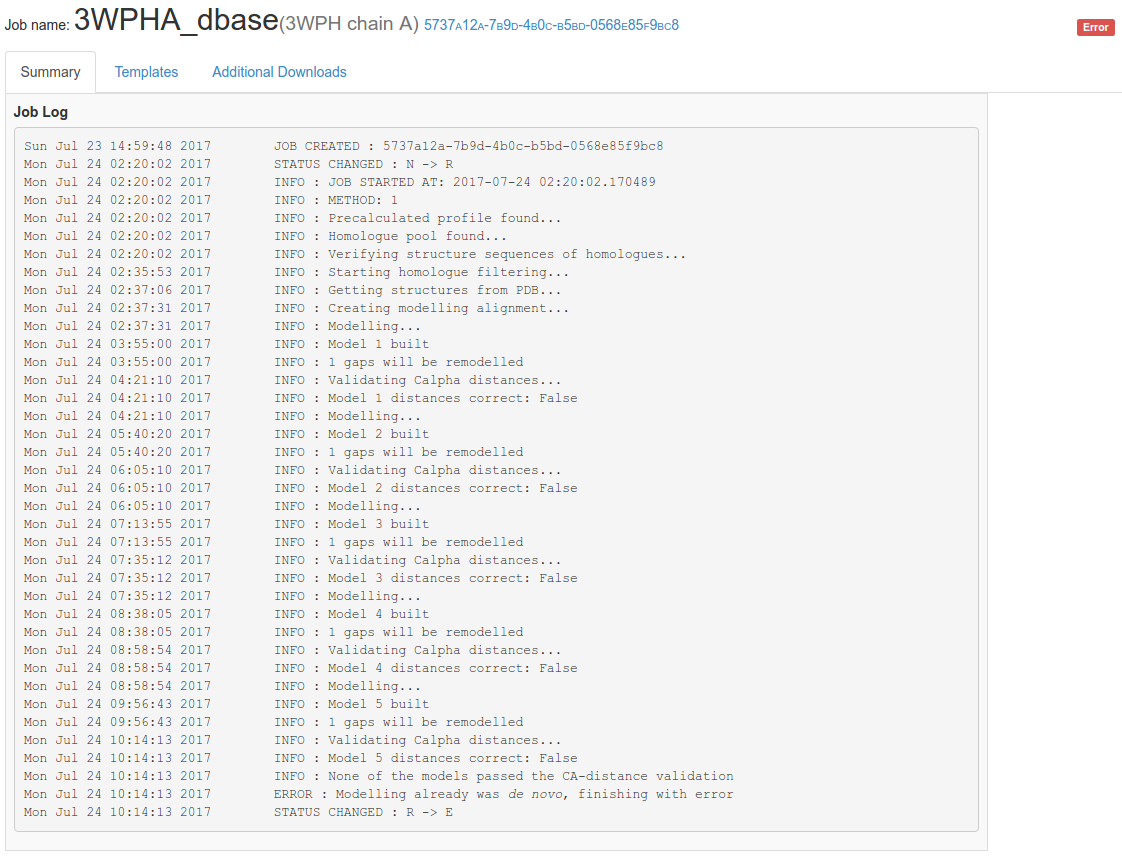
Fig. 2 Example of failure: Job log of modelling gone wrong.
If the modelling has been completed (e.g. only the entanglement detection failed) all the information calculated prior to the failure is available (all the panels described in General information section).
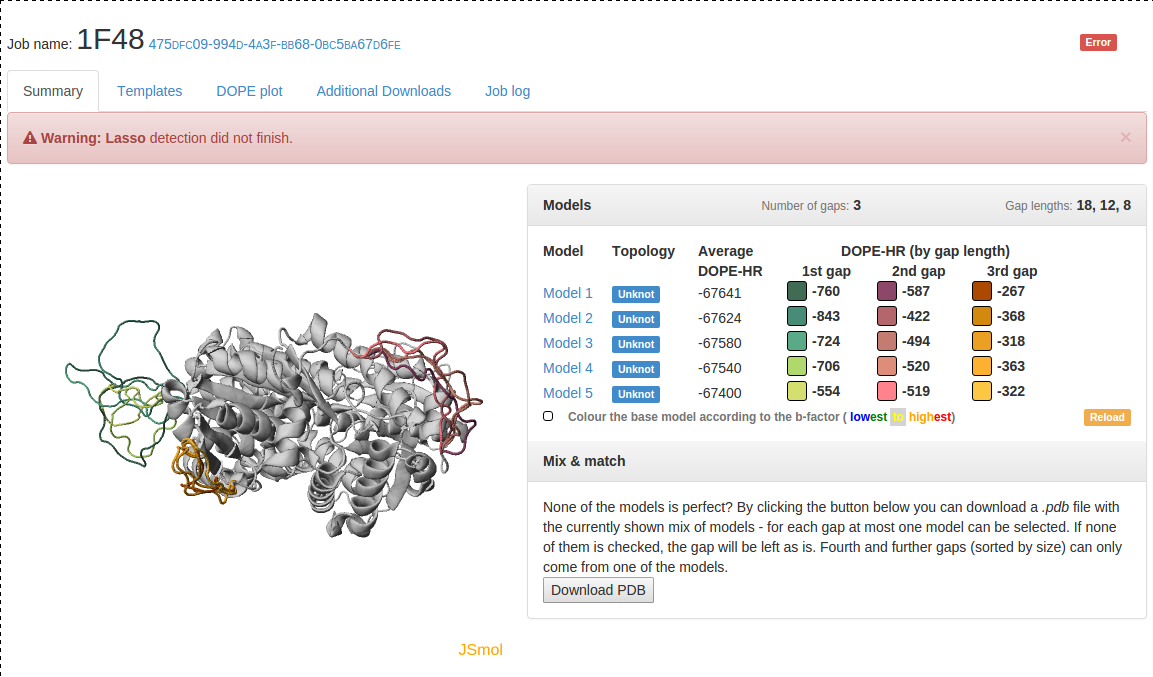
Fig. 3 Example of failure: Modelling went without a hitch, but some problems were encountered during entanglement calculation.
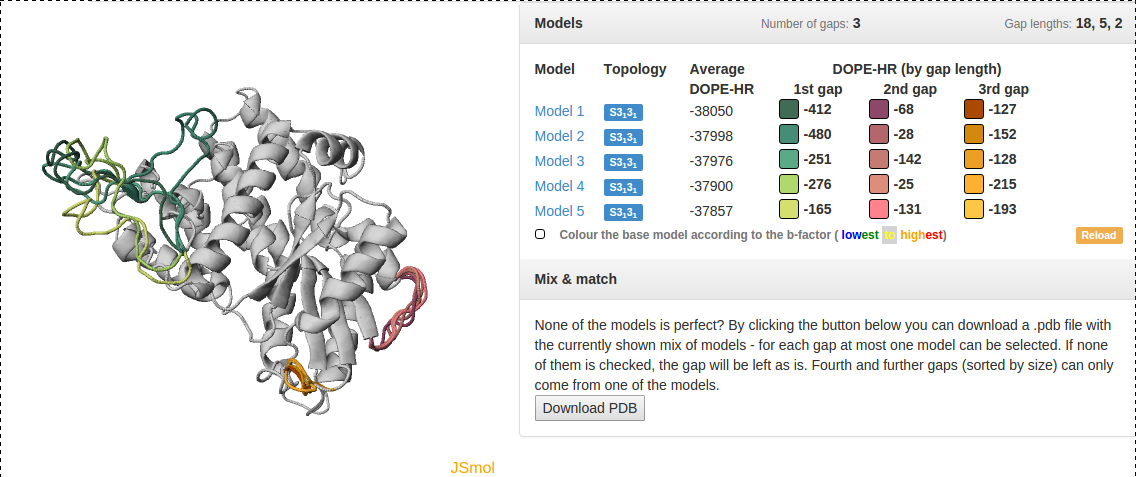
Fig. 4 An example of data presentation for a knotted protein with PDB ID 1ual repaired using GapRepairer. Applet in top left: graphical representation of protein in JSmol. Top right: DOPE-HR scores for each model/gap combination and control panel for applet. Bottom right: Download button for a structure created from multiple models.

Fig. 5 An example of structure visualization for a knotted protein with PDB ID 4mcb repaired using GapRepairer. Left: Electron density map (σ=1, 1.5Å around the structure) overlay. Right: Difference electron density map (σ=3, 3Å around the structure) overlay.
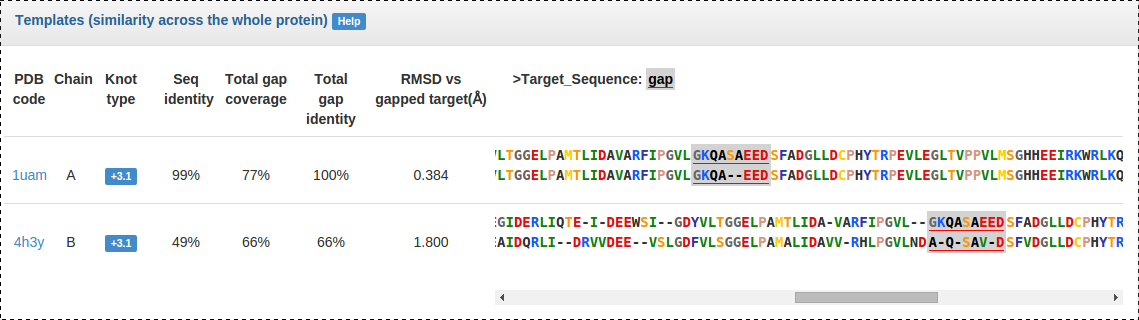
Fig. 6 Templates panel: statistics pertaining to each selected template and pairwise alignment to the target. Amino acids in the alignment coloured according to the scheme used in I-Tasser. Modelled gaps indicated by a gray highlight, with three largest gaps underlined additionally in red.
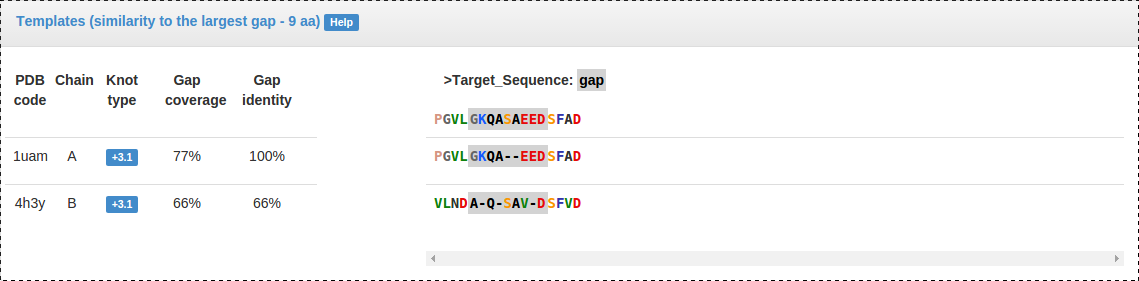
Fig. 7 Gap panel: present for each gap being repaired, specifies gap coverage and identity for each template, and the multiple sequence alignment used for homologous modelling.

Fig. 8 Excluded structures: panel which lists proteins excluded by the user - not necessairly proteins which would be used if not for this exclusion. Syntax mirrors the one used in the input field - if chain was specified it is added for a given PDB ID after the colon.

Fig. 9 Download tab: links to model files and alignment used for modelling.
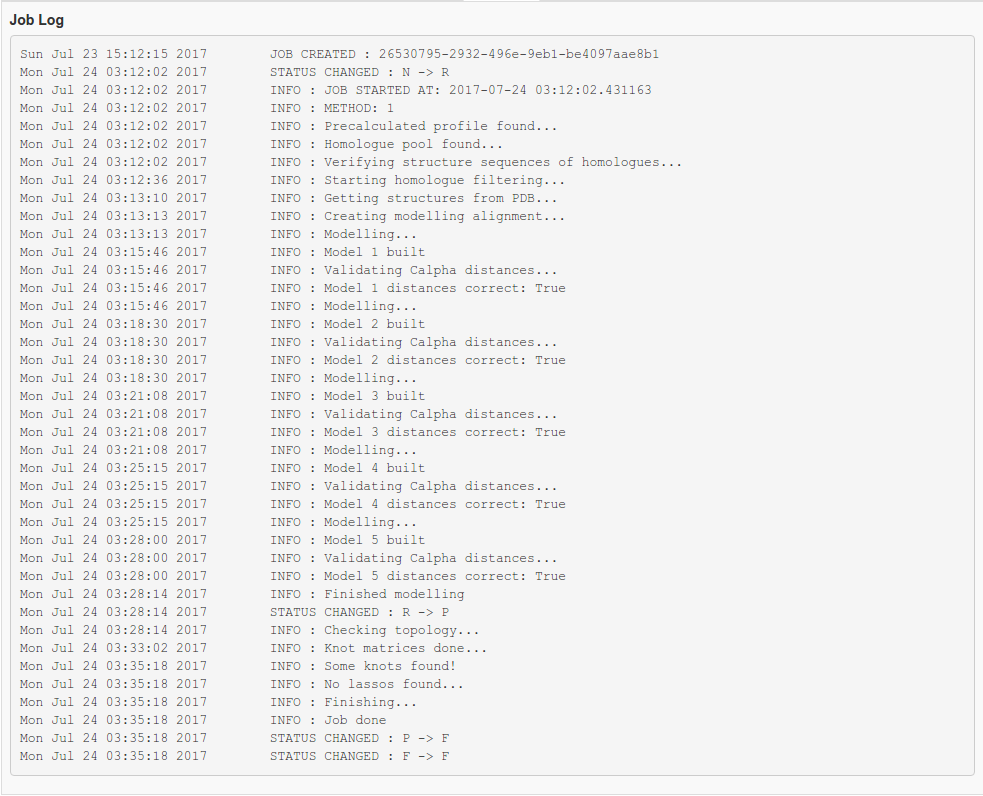
Fig. 10 Job log.
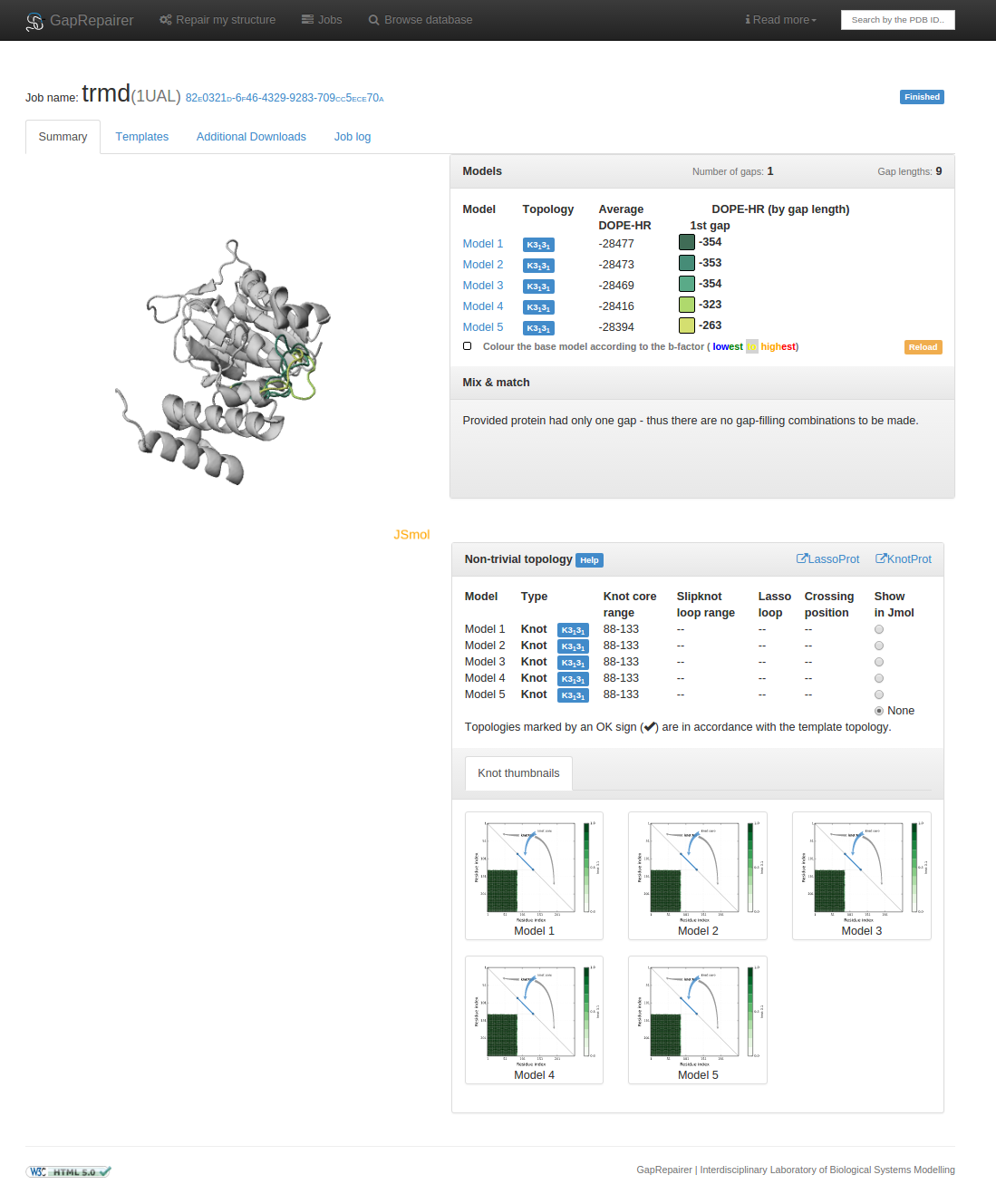
Fig. 11 An example of data presentation for a knotted protein with PDB ID 1ual repaired using GapRepairer. Applet in top left: graphical representation of protein in JSmol. Top right: DOPE-HR scores for each model/gap combination and control panel for applet. Middle right: detailed data about knots/slipknots and lassos formed by backbone subchains. Bottom right: diagram presenting a topology matrix for the models.
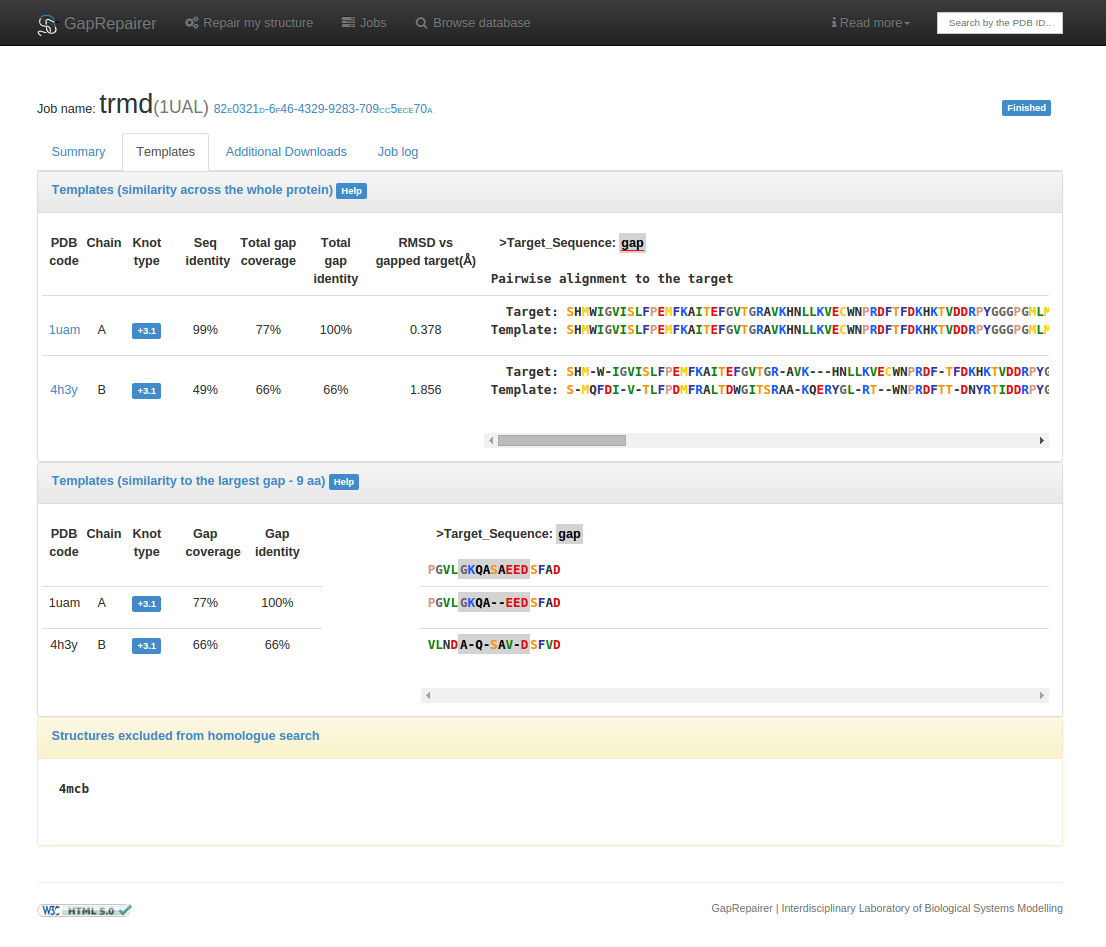
Fig. 12 An example of selected homologues presentation for a knotted protein with PDB ID 1ual repaired using GapRepairer. First panel shows averaged statistics for each template based on a pairwise sequence alignment with target. Middle panel presents multiple sequence alignment used for modelling missing residues. Last panel lists structures excluded from the homologue search (in this case: all chains form the protein with PDB ID 4mcb).
For protein chains with any kind of entanglement additional panel is available on the summary page:
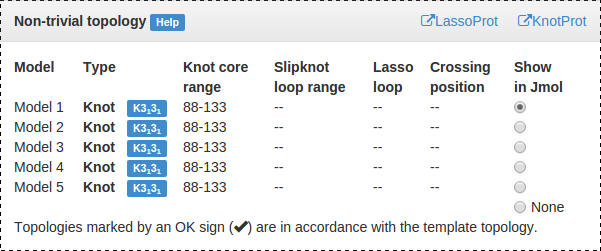
Fig. 13 Table detailing location of the outmost knot/slipknot for each model. Radio button in the last column displays this entanglement in the JSmol applet, and enlarges and highlights the corresponding matrix.
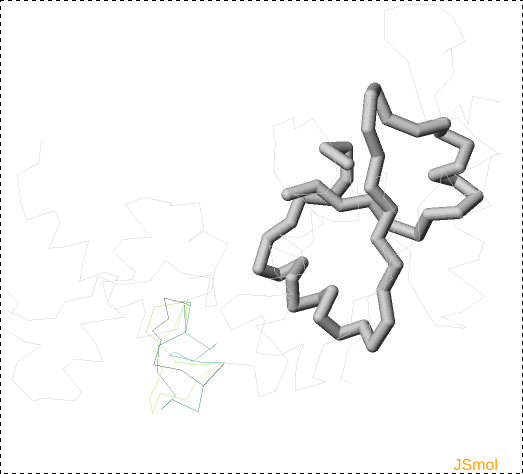
Fig. 14 Knot location visualized in the JSmol applet: knot core (chain segment that elongated by one aminoacid would already form the knot) is show as the thickest line coloured blue, slipknot loop (not present in this picture) has medium thickness and yellow colour, and rest of the protein is a thin grey line.
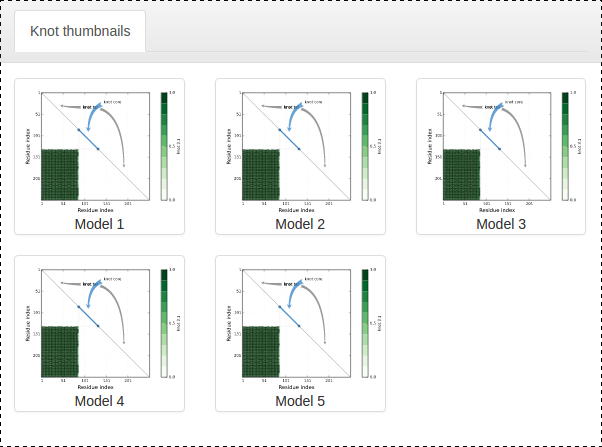
Fig. 15 Topology matrices which indicate presence (or absence) of a particular entanglement, were the chain cut only to the segment between given position (eg. if position (35,160) is coloured green chain contains a 31 knot somewhere between 35th and 160th amino acid).
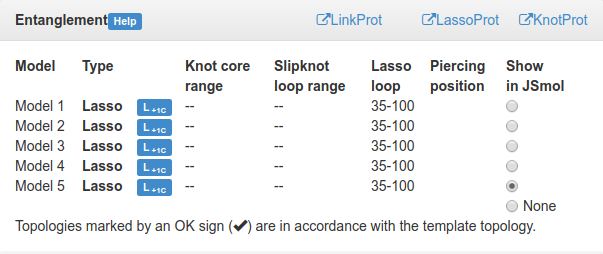
Fig. 16 Table detailing location of the outmost knot/slipknot for each model. Radio button in the last column displays this structure in the JSmol applet, and enlarges and highlights the corresponding matrix.
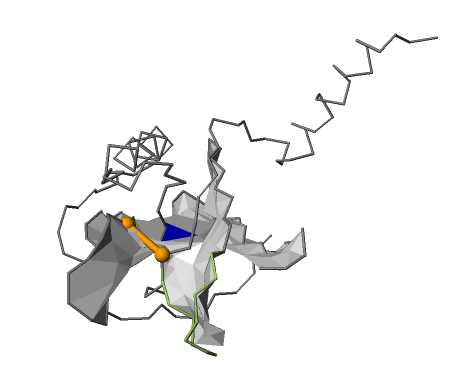
Fig. 17 Lasso location visualized in the JSmol applet. Cystein bridge that closes the lasso loop is imagined in yellow, surface of the loop is formed by gray triangles with the crossing coloured blue.
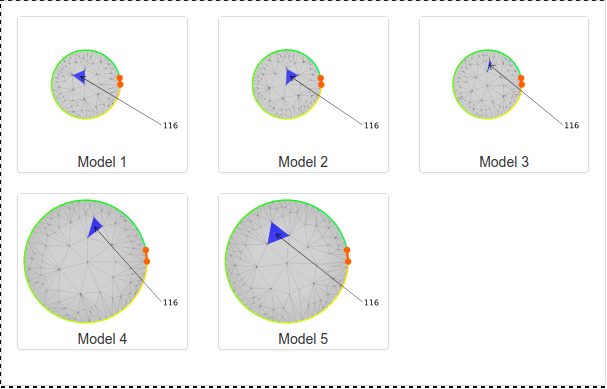
Fig. 18 Outmost lasso loop of a model visualized as a circle, with the crossings marked in blue and annotated by the number in the protein chain of the amino acid closest to the crossing on the threaded fragment.
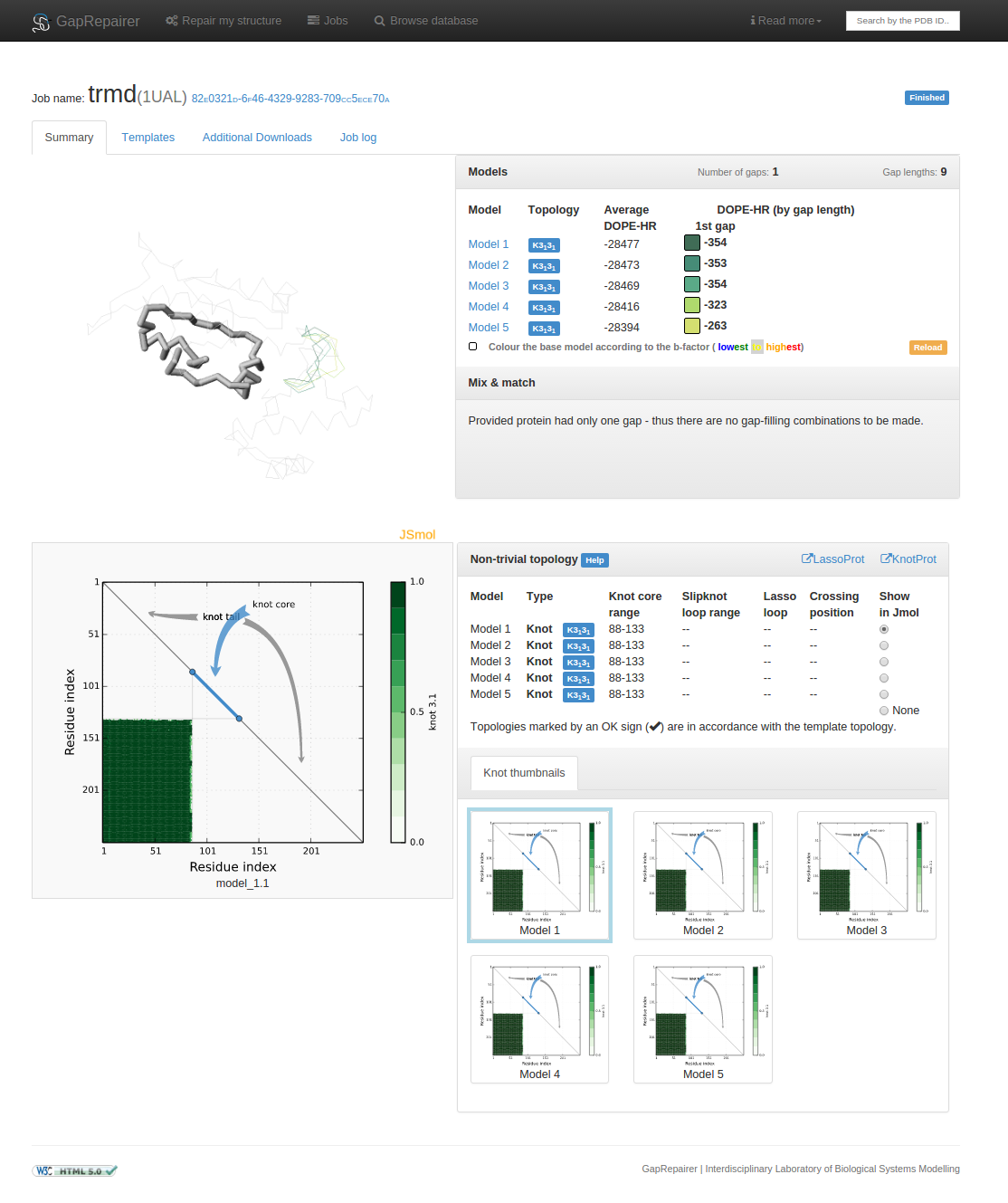
Fig. 19 An example of data presentation for a knotted protein with PDB ID 1ual repaired using GapRepairer.
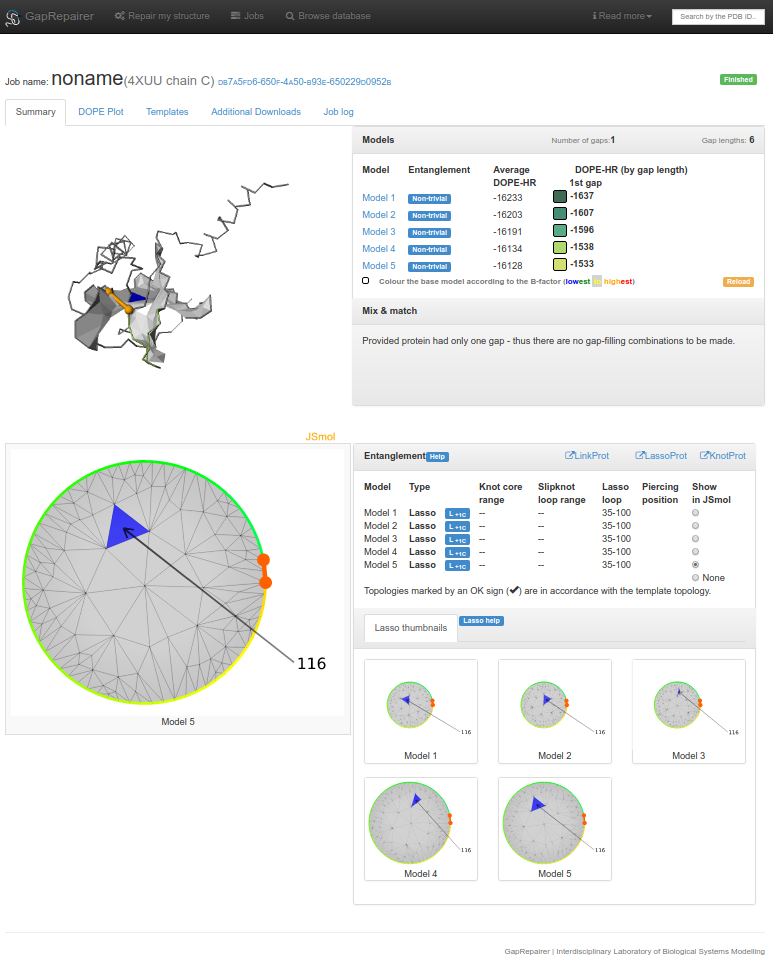
Fig. 20 An example of data presentation for a protein with PDB ID 4xuu (chain C), which contains a lasso, repaired using GapRepairer.
 GapRepairer
GapRepairer