Due to experimental limitations, over 25% of protein structures possess unresolved fragments. This means that up to a quarter of works in drug discovery, protein interaction etc. may be slowed down, because researchers do not have appropriate tools to model missing parts of protein chains. Currently existing methods are either sophisticated bioinformatical packages (e.g. Modeller [1]), or over-simplified servers lacking many important capabilities. Moreover, no existing method takes into account the topology of the target (existence of knots [2], lasso structures [3] etc.), although the topology may be decisive for the function and properties of the protein [4].
In this work we present the GapRepairer – the server that fulfills the gap in the spectrum of structure modeling methods. The server, by its attractive and intuitive interface, offers the power of Modeller homology modeling even to unexperienced users. Moreover, both template selection and final model validation enable detection and analysis of knot and lassos.
To submit the structure for repair, it is enough to enter its pdb code, or upload the (gapped) file with atom coordinates (pdb file) along with appropriate protein sequence in fasta format. The gaps are identified automatically, as well as the appropriate templates for homology modeling. To reduce disagreement between various templates, only those with entanglement compliant with the best homologue are kept. The best predicted models are shown superimposed using JSmol presentation (Fig. 1). This allows for visual inspection of obtained models using standard JSmol options (rotate, zoom, etc.). For the proposed models complex topological analysis is also conducted. In particular, both knot/slipknot matrices [2] and lasso presence [3] are computed and presented.
Apart from topological filters, the GapRepairer server has additional features, unique among web-servers. The modeling is done with as few restrictions on possible targets as possible, e.g. gap count, and length, are limited only by template availability, while in most servers only one gap can be modeled. Furthermore, users can easily mix obtained models by choosing different gap fillings from different models. Moreover, while to date only one chain can be repaired at a time, to avoid clashes users can choose to take into consideration other chains present in the file. The calculated models are sorted according to their homology assessment (dope potential), which is also presented as a plot in a separate tab. Users can also analyze the templates and the alignment in respective tabs.
As using Modeller requires license from every user, when this license is missing GapRepairer prepares downloadable files (which include both cleaned template files and filled-out python script), which allow users to easily run the modeling on their own machine after obtaining Modeller license.
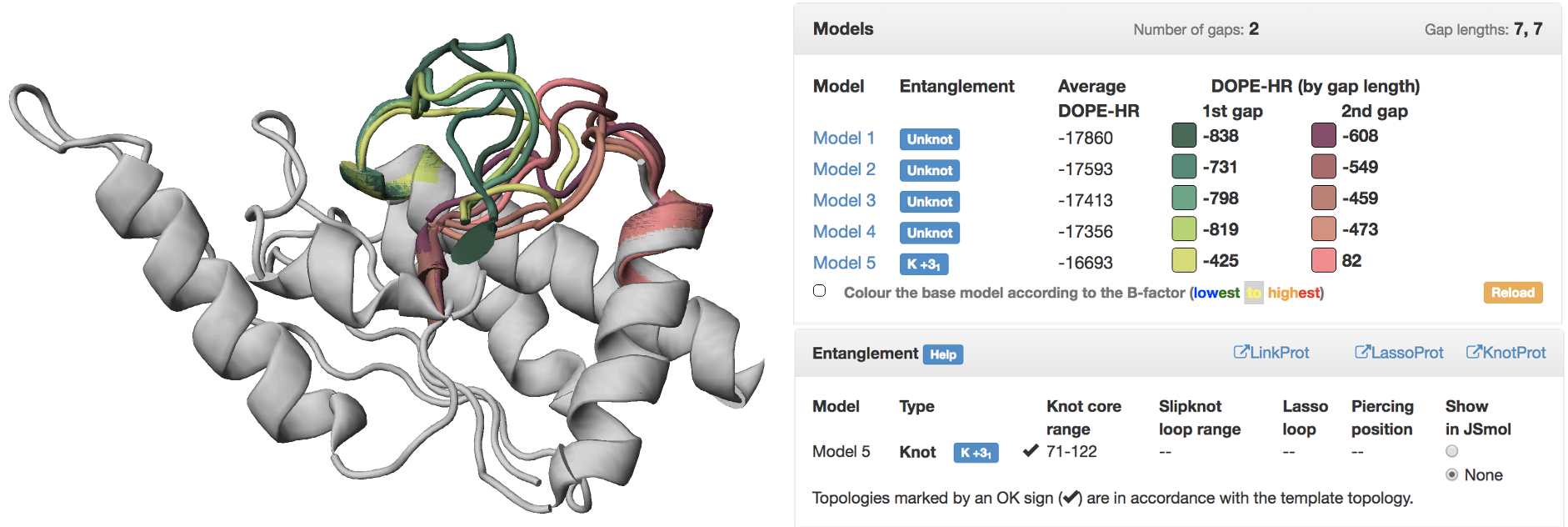
Fig. 1 Exemplary output of the modeling. On the left the uploaded structure (gray) with filled gaps (color loops). The colors correspond to the models in the table on the right. In the top table 5 best models are presented along with their topology and DOPE potential. The display of the model in the JSmol presentation may be turned on/off by clicking on appropriate color box. In the table below the details concerning nontrivial topology are shown.
For more advanced users, the GapRepairer server offers many adjustable options. In particular, users can include or exclude chosen structures, change cutoff e-value for template search, decide on alignment building or perform homology search across the structural (not sequential) homology database (DALI [5]), which is a novel feature. The server may also be invoked from the command line, which may be used in automated gap filling for large set of proteins, e.g. during the CASP competition.
To test the performance of the server, GapRepairer was compared with standard methods for loop modeling ( Rosetta [6], CABS [7]), achieving comparable, or even better results. The topology of modeled structures was also checked in a blind test – over 98% of repaired structures were modeled with proper topology. Therefore, the GapRepairer was successfully used to repair incomplete structures found in topological protein databases KnotProt and LassoProt [2,3]. The results form a unique database at the server webpage (where currently over 120 proteins can be found, with this number still growing). We strive to repair and share as many of the chains deposited as artifacts in topological databases (like KnotProt), as possible.
Most of similar servers use de novo modeling, starting from the sequence only (e.g. I-Tasser [8], Swiss-Model [9], HHPred [10] etc.). As a result, the details of resolved parts are not taken into account and the whole calculation lasts much longer. The servers devoted only to loop modeling (e.g. ArchPred [11]) model only one loop, giving users no sufficient freedom to adjust parameters, and do not check the topology (knots or lasso) of predicted models. In particular, no server known to us allows to search through structural (not sequential) homologs database.
Because of its intuitiveness and versatility, the server will be useful for broad spectrum of researchers. The server is currently used by many groups.
 GapRepairer
GapRepairer