Workflow of GapRepairer can be divided into several steps:

Fig. 1 Workflow of Gap Repairer. Each vertical pathway shows steps performed by the server given a particular option chosen by the user.
To repair a structure present in the RCSB Protein Data Bank database the only thing needed is its PDB ID (e.g. “1UAK”) and which chain is to be modelled. Chain is a case-sensitive (following the PDB file format specification) and always consists of only one letter/number. Only residues with this symbol at the 22nd column in the .pdb file will be part of ther target for modelling. Gaps are found based on a pairwise alignment of expected sequence and structure-read sequence as deposited in the database – thus a chimeric protein will be seen as complete, as long as no ATOM records are missing. By default only gaps within a sequence are repaired – uncrystalized segments that are not preceded AND followed by known residues are trimmed. To rebuild this fragments also "Model missing tails" option should be selected (under "Show advanced options").
Both structure and sequence files will be created based on .cif files available online at PDB. Then only exception are so-called "Large structures" which contain to many atoms/residues/chains to be correctly formatted into a PDB format. In such a case, you should upload the chain in question (optionally with its neighbours) using the second input option. Non-protein HETATM atoms (such as belonging to ligands) are removed from the structure based on information available with RESTful services from RCSB PDB
To allow repairing of structures not present in the Protein Data Bank database it is possible to upload a .pdb file. Such a file must be correct according to the .pdb file specification and contain
ATOM or HETATM records for all residues with known positions. It is important that the 22nd column of residues to be taken into consideration while modelling must contain the symbol specified in the “Chain symbol” input field (if no chain symbol appears in the
structure file, corresponding field should be left blank, or contain one
Uploaded structure file will be edited to remove probable non-protein entities (such as ligands). For atoms with different possible positions specified in the .pdb file only their first appearance in the file will be kept.
Since .pdb files do not necessarily contain information about uncrystalized residues a .fasta file with the full expected sequence of the protein must also be uploaded. This file will be used to determine position of the gap(s) and what it should be filled with. Only gaps within a sequence are repaired – uncrystalized segments that are not preceded AND followed by known residues will be trimmed, unless the "Model missing tails" option is selected.
Only structures which can be expected to result in a valid model are accepted. This filtering is made based on a lax distance criterion - straight-line distance between consecutive C-alpha atoms should not exceed 5Å. For uncrystallized fragments this means, that for each 5Å on the straight line between gap flanking residues, there should be at least one residue in the fasta sequence of the gap.
Default template selection methods are sequence based – sequence of the protein to be repaired is run using locally installed PSI-Blast against a monthly updated PDB database copy (to reduce wait time most of the published structures have similarity profiles already precalculated). Default e-value cutoff, after which sequences are discarded is set at 0.001, but can be changed under “Advanced options”.
Structure files of selected proteins, as well as their full sequences, are downloaded from the RCSB PDB database following the procedure described in the "Input structure selection" - "PDB Code" section.
Structure-based template search is currently implemented only for published structures and consists of a PDB ID search against DALI database. Dali Z-score cutoff used for first filtering of the structures is set at 2 (indicated by method creators as significant similarity).
Structure files of selected proteins, as well as their full sequences, are downloaded from the RCSB PDB database following the procedure described in the "Input structure selection" - "PDB Code" section.
It is possible to specify particular structures to be used as templates. When PDB IDs (which may also include a chain specifier) are given by the user, their structure files are downloaded from the RCSB PDB database following the procedure described in the "Input structure selection" - "PDB Code" section.
It is also possible to upload structure file from the hard drive. This file should meet all the same guidelines as the uploaded target structure (accordance with .pdb file specification). In particular it should be noted that in case of any missing residues in the uploaded template the resulting alignment to the target may not be optimal.
In some cases it may be better to remove some structures from the potential template pool (eg. when rebuilding a published structure – it would obviously be the best match for itself). PDB IDs of structures to be disregarded in the template search should be specified in the “Structures to exclude” input field under the Advanced Options tab.
Main foundation of our server, which inclusion separates GapRepairer from other currently available gap-filling protocols, is that the topology is highly conserved. What follows, is that even distantly related proteins, with little sequential similarity, tend to keep within the same class of topological structure [1]. We revisit this idea in the “Template selection” section below, as topological filtering is an important step in final template determination. Read more...
Topology in proteins is defined as a presence (or absence) of knot- or slipknot-like structure (we will refer to them further as just knots or slipknots but it should be noted, that proper, mathematical definition of such structures involves a closed chain, obviously missing in the proteins). Knots and slipknots are referred to using numbers (like 31, 52), with the larger one indicating number of crossings present, and the index given to differentiate between differently folded structure with the same number of crossings. Fig. 2 below shows the mathematical 31 knot (left) and the same knot "relaxed" to better fit the protein knots shown in neighbouring images (right), with the implicit connection of protein chain ends indicated by a dashed line.

Fig. 2 Trefoil (31) knot. Right hand side image relaxed to a postion more similar to proteins' in Fig. 3 and 4 (chain closing segment, which does not appear in proteins indicated by a dashed line).
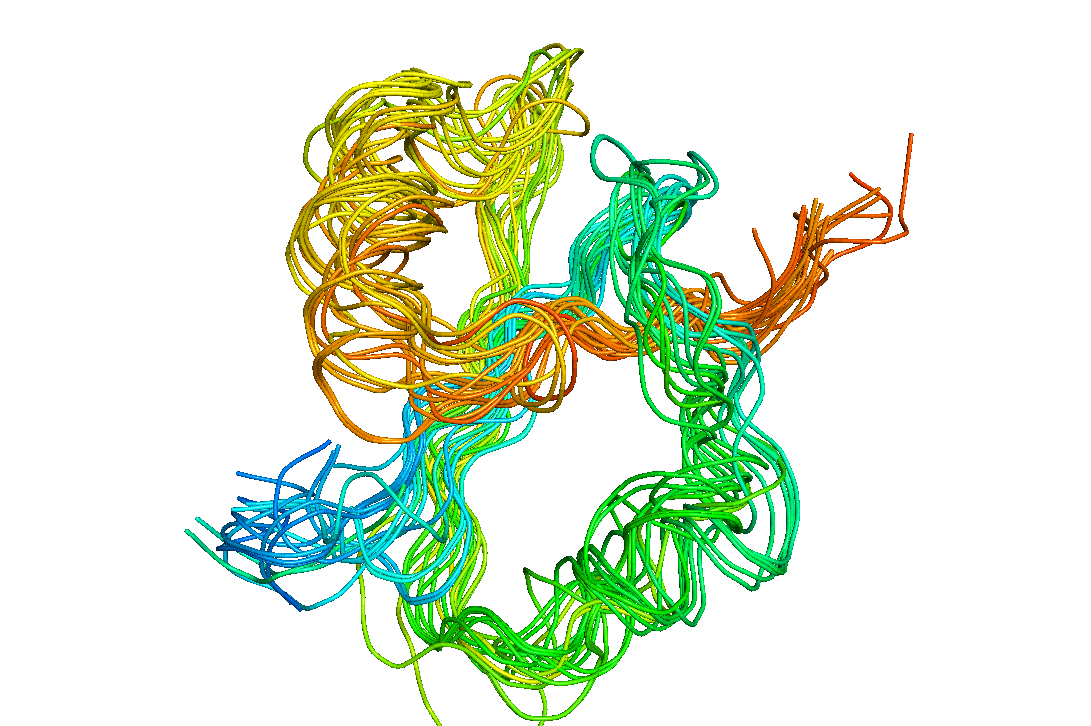
Fig. 3 Structural superposition of knots belonging to various methyltransferases.
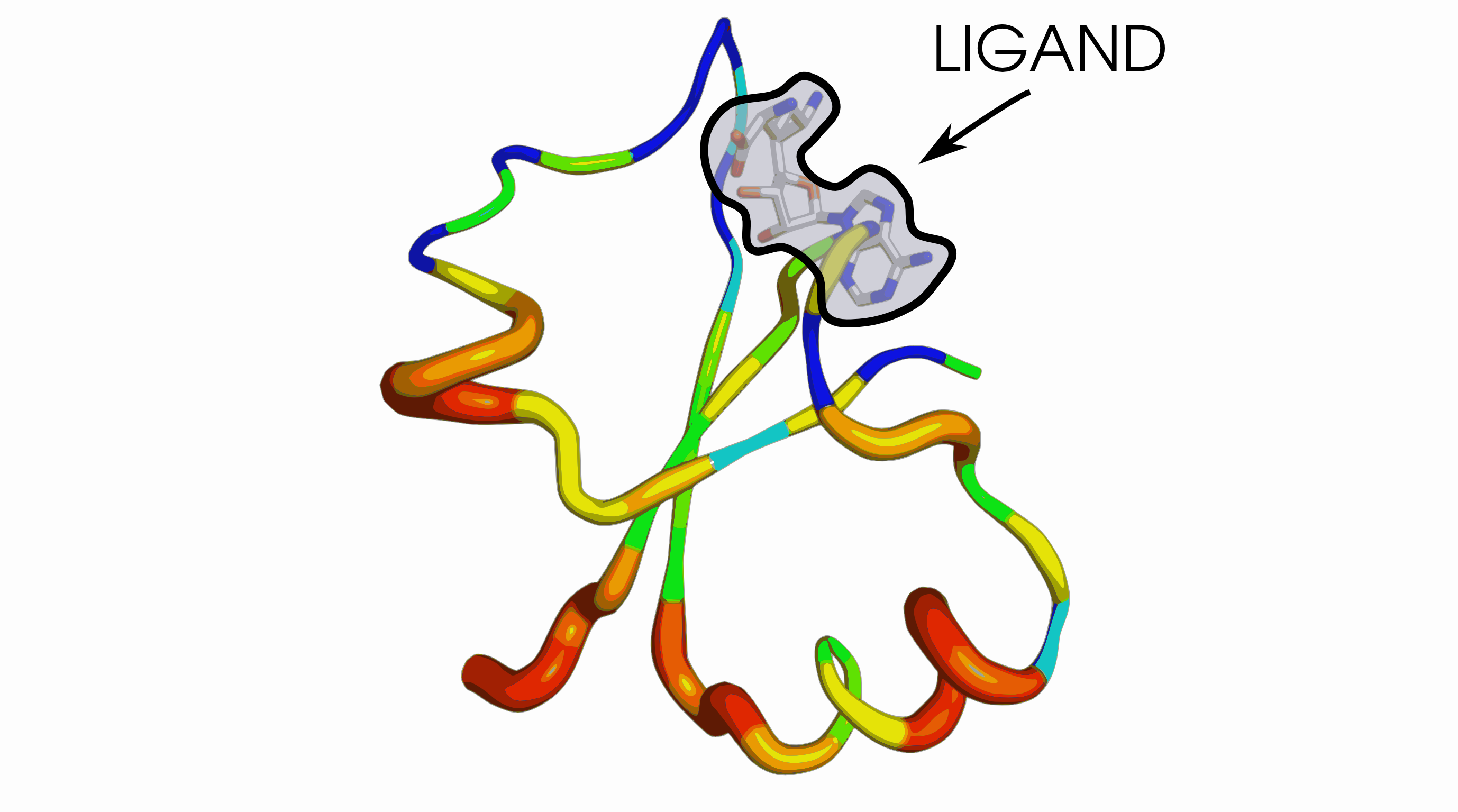
Fig. 4 Shannon variablity of the multiple sequence alignment of knot sequences from a TrmD family, mapped onto a representative knot (from structure with PDB ID 4MCB, chain B). Increasing thickness and colour (from blue to red) indicates increasing entropy.
Fig. 3. above presents a structural alignment of the 31 knots found in various members of the methyltransferase enzyme class (not necessairly belonging to the same protein family). Fig. 4. visualizes Shannon's entropy of just one of the methyltransferases families on one of the structures (with thickness indicating increased variability of the given position). As can be seen only the binding site is conserved, yet even with high sequential diversity of the rest of the fold, 31 knot present in the aligned proteins still remains nearly the same.
Further information about nontrivial topologies encountered in proteins can be found at KnotProt database and server.
All potential templates (taken from either sequential or structural search) are pairwise aligned to the target sequence and then sorted based on the resulting
sequence_identity_within gap * sequence_coverage_within_gapscore, averaged across all gaps (with gaps expanded by 4 amino acids in each direction to ensure better fit). The highest scoring templates is then taken as a reference for the topology filtering. Topology of each potential template is taken from the KnotProt database and only proteins matching the reference topology are kept.
Restriction of the templates to only one topology is important, as it may influence the topology of the final structure - if the gap is within the potential knot, different ways of filling this gap may result in unexpected crossings of the chain. Eg. when we want to correct the topology of the methyltransferase with PDB ID 1OY5 (unknotted, likely as an error considering its close homologues) addition of unknotted structures into the template alignment may result in the starting model being also unknotted. By excluding the original structure from the search, and restricting the topology to the then most similar homologue's, we can enforce the prefered topology. Proteins 1OY5 and its closest homologue 4mcb are superposed in Fig. 5. More details on this case can be found in the Possible uses section.
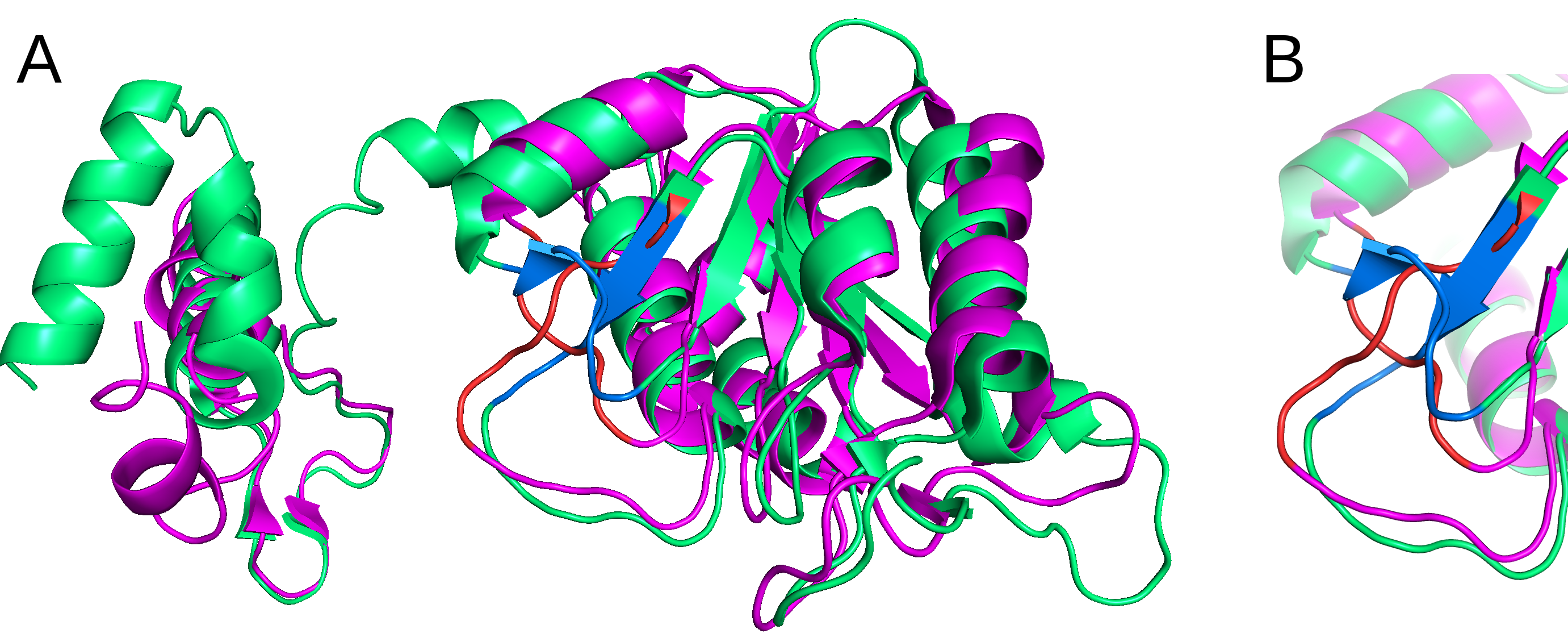
Fig. 5 A Superposition of a knotted (PDB ID 4MCB, chain B, shown in green and blue) and, erroneously, unknotted (PDB ID 1OY5, chain A, shown in pink and red) proteins. Malformed crossing coloured blue and red are shown in the panel B. B Closeup on the malformed crossing (coloured blue and red) from panel A.
For the “Only one – the best one” Homologue selection method only this reference sequence is returned.
When multiple templates are to be used their numbers are further reduced to ensure that only the necessary minimum is used. To this end sorted templates are analyzed one by one against the current alignment. A template can be marked as useful for a given gap if it has at least a 30% coverage of the gap region, and it covers some residues which has not yet been covered by previously found templates. This continues until full coverage of the gaps is reached (or all potential templates have been exhausted).
When templates are specified by the user the above procedure also applies, except potential template pool is restricted to user-defined structures and no cuts are made based on similarity e-value.
Due to the specific requirements of the MODELLER software used, and to ensure that the known part of the structure is not moved during the modelling process, templates are only aligned to the target sequence within gaps. Gap-filling sequences are determined based on the pairwise alignment to the target and then a multiple sequence alignment is created, independently for each gap.
Gaps shorter than 2 residues, and sequence tails where specified, are modeled de novo.
Based on the alignment prepared in the previous steps up to 5 models with filled gaps are created using a modified AutoModel class form MODELLER package.
This class uses template-derived restraints as a starting point (with the modification freezing rest of the protein to stop any changes) – a consensus structure that averages positions of the residues corresponding in the alignment to each of the missing amino acids.
Such created structures are then refined through molecular dynamics based annealing with
md_level = refine.very_slow library_schedule = autosched.slow max_var_iterations = 500 repeat_optimization = 3 max_molpdf = 1e6. Only residues selected for optimization can be moved during this process - these include all the gap regions, residues for which some of the atoms were missing, and tails, broadened by the flanking residues on both sides. Each gap within the model that has DOPE above 1000 (which is very bad) is then rebuilt with a specialized loop optimization class
LoopModel. Out of the original loop and 5 new attempts only the best scoring, and containing valid distances, variant is kept.
Each model is then validated to ensure proper Cα distances, and scored using the DOPE-HR method implemented in the MODELLER. Each model that couldn't pass the distance validation is then rebuilt de novo (if it wasn't yet) and once again validated.
Only models with all consecutive Cα distances below 5Å are kept.
The knots and slipknots formed by the chain are detected according to the method introduced in [2,3]. As the proper, mathematical knots are defined only for closed curves, the crucial step in the method is chain closing. It is being done by projecting the ends of the chain randomly on the edges of C60 fullerene large enough to contain whole protein (Fig. 6). The chain is afterwards closed on the surface of the fullerene. For the closed curve, one can compute the Alexander polynomial, which is knot invariant distinguishing between simple kinds of knots.
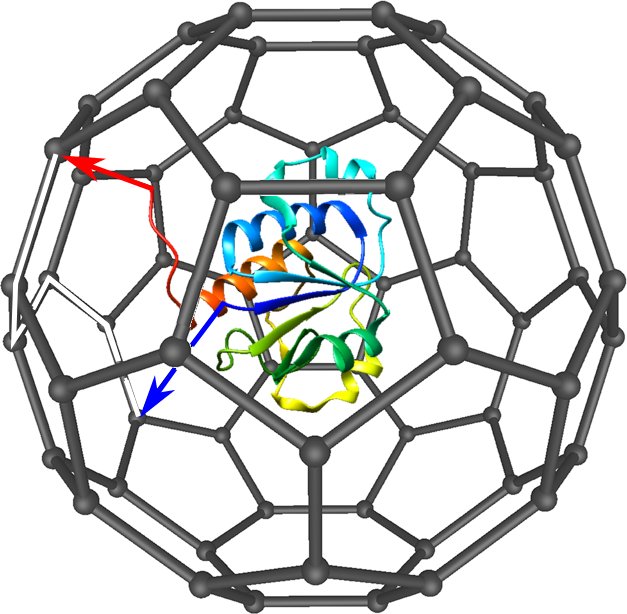
Fig. 6 Scheme of used chain closing method. The protein is caged in a C60 fullerene, the chain termini are randomly projected on the edges of fullerene (black arrows). The chain is closed on the fullerene surface (white track).
In general, the knot type can depend on the projection direction (especially when the chain termini are hidden inside the protein). Therefore, to decrease the projection direction effect, large number of projections (1000) is being done. The knot is considered to be real if at least 42% of projections revealed the knotted topology of a protein.
The topological analysis is crucial in homology modelling. Our results show, that correct arrangement of the chain is achieved only with the use of homologs with proper topology [4]. The GapRepairer is to our knowledge the only server taking care of topologically consistent modelling.
Except for knots, the server analyzes also the existence of slipknot topologies. The slipknot topology is formed e.g. by the shoelaces. In other words, the slipknot arises, when the part of the chain would be considered as knotted, however the chain as a whole is not knotted.
To detect slipknot topologies the server conducts the same analysis as for knot, however for shortened chain. As the chain can be shortened both from N- and C-terminus, finally one obtains a matrix (Fig. 7), encoding the topological state of the protein [1]. In particular, the spot in the matrix with coordinates (i,j) denotes the topological state of a piece of the chain from i-th residue to j-th residue.
Such approach enables in particular to define the knot core (the minimal part of the chain, which can be still considered as knotted), knot tails (the part of the chain outside the knot core), slipknot loop and slipknot tail (Fig. 7).
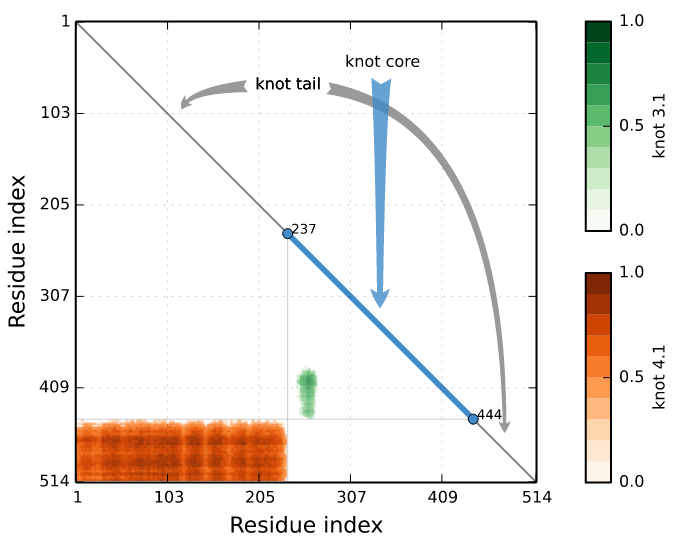
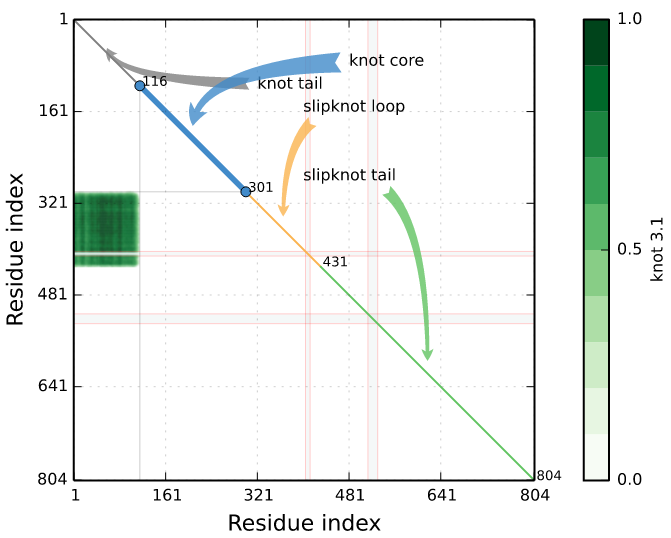
Fig. 7 Exemplary of knotting fingerprints (matrix diagram) detected by Knotprot in proteins (PDB IDs 5E4R chain A and 5JIS chain A). The left panel shows complex topological fingerprint denoted as K4131, which is composed of two types of entanglement (the 41 and the 31 knot). Right panel shows a slipknot, which is composed of the 31 knot). The bars in the right of each matrix denote the probability of obtaining each knot type.
The right panel shows an example of the matrix diagram constructed for a protein with missing atoms. The missing atoms are shown by grey strips.
When grey strips are located in the knotted core of protein, and the missing part of the chain is replaced by a line (straight) segment this may affect the type of knot detected. In these cases we recommended to use GapRepairer to carefully reconstruct missing segments taking into account topology of know homologous proteins. An example of the knotting fingerprint of protein after reconstruction with GapRepairer is shown in the Figure 8.
The concatenation of knot and slipknot types forms a knot fingerprint – the highly conserved notion characteristic to protein topology [1].
In fact, many knotted and slipknotted proteins possess a gap. E.g. in the matrix depiction in Fig. 7 there are straight lines denoting the missing fragment. The goal of Gaprepairer server is to fulfill this fragments obtaining correct topology (which sometimes can mean change of structure topology). Fig. 8 presents the matrix fingerprint of human autotaxin inhibitor before and after gap modelling.
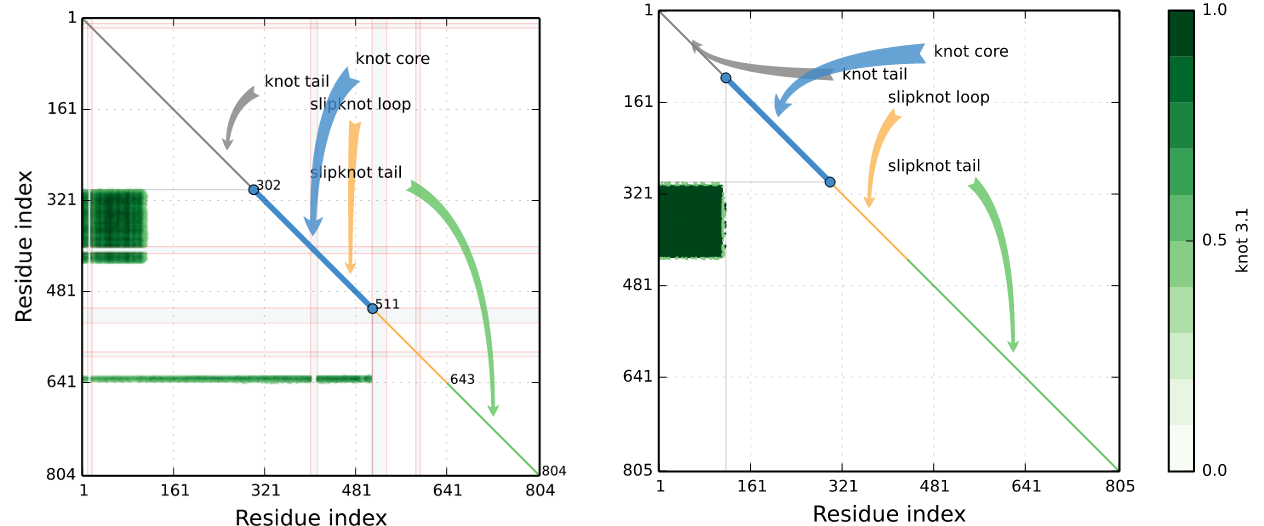
Fig. 8 Knotting fingerprints of a protein (PDB ID 4ZG6 chain A), constructed before and after gap filling with GapRepairer.
The commonly occurring in proteins disulfide bridge closes a chain forming a covalent loop. In such case it is possible, that a chain terminus pierces through the loop. Such configuration is called complex lasso protein [5,6].
The lasso configurations are detected applying the method introduced in [4]. The crucial part of the method is spanning a surface (triangulation of minimal surface) on the covalent loop. Afterwards, the piercings are calculated, as the crossing of the chain with a surface. The orientation of the surface allows to prescribe also the direction for each piercings. The piercings which are too shallow are reduced (Fig. 9). Finally, one obtains the lists of (signed) piercings through the surface. This allows to prescribe for each loop a lasso type, characteristic for protein families [6].
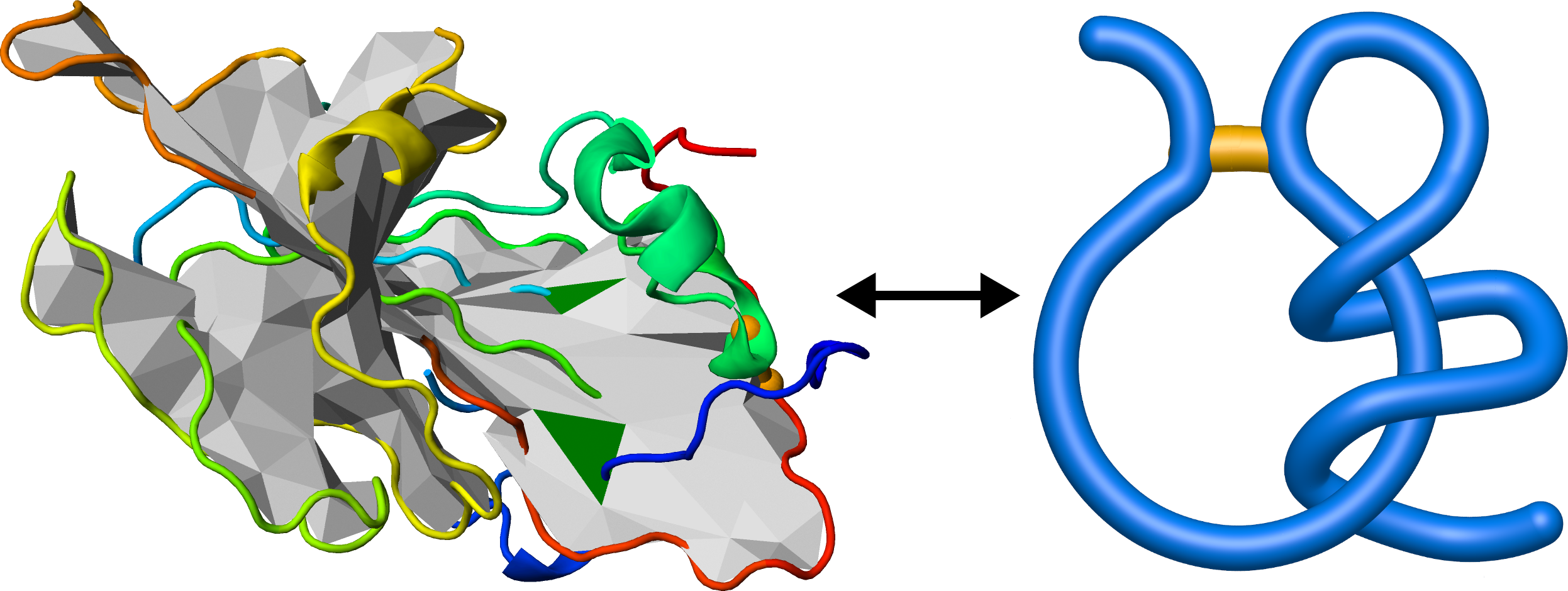
Fig. 9 Example of lasso analysis (left panel). The surface spanned on the chain (grey) is pierced by the blue parts of the chain through green triangles. The lasso tail winds around the covalent loop forming a supercoiling lasso motif (right panel).
If in protein there are more than one loop, the concatenation of lasso type symbols forms a lasso fingerprint [7].
As the lasso topology is in general conserved among homologs, the lasso type can be a pivotal factor in structure assessment [6]. In particular, such analysis was used to find wrongly determined parts of structures [8].
In our server disulfide bridges are determined geometrically by MODELLER – that is all cysteines with sulfurs less than 2.5 Å apart are treated as a potential bridge.
 GapRepairer
GapRepairer